Rapid Exploration of the Assembly Chemical Space of Molecular Graphs
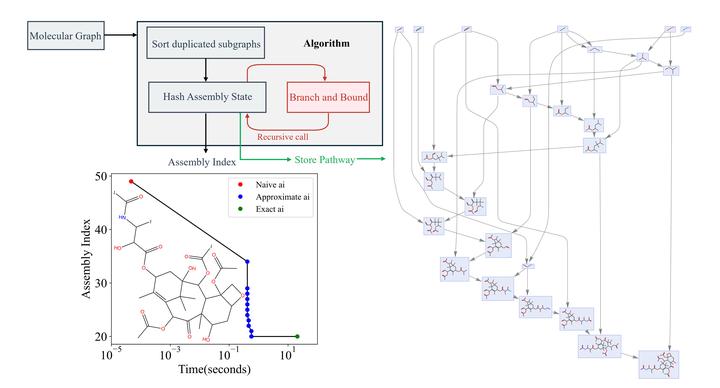 Journal Paper Figure 4,5: Algorithm pipeline and output
Journal Paper Figure 4,5: Algorithm pipeline and outputAbstract
Quantifying how hard it is to build a molecular graph matters for biosignature detection, chemical complexity, and cheminformatics. We present an exact, scalable algorithm to compute the molecular assembly index (MA), which prioritizes the largest duplicate subgraphs, represents fragmentation with an array of edge-lists, and prunes the search with both dynamic programming via a hash table of assembly states and a branch-and-bound heuristic guided by a conditional addition-chain lower bound. For organic molecules in the greater-than-500 Da range, our approach is up to 6 orders of magnitude faster than prior methods and yields exact MAs where previous algorithms would have timed out. We compute MAs to convergence for ∼300k COCONUT natural products with <50 bonds, profiling time and memory scaling. Finally, we exploit the speed of our algorithm to calculate joint assembly spaces and introduce the Joint Assembly Overlap (JAO), a Jaccard-like metric that emphasizes global scaffold reuse, and show that the JAO yields substantially different rankings from Tanimoto similarity with ECFP fingerprints and MCS (e.g., in steroids 270–380 Da and short peptides), accounting for substructural similarity beyond local environments. Together, these advances turn the molecular assembly index into a practical tool for large-scale exploration of chemical space.
Keith Y. Patarroyo
Research Fellow on Chemical Intelligence, Open-Endedness and Hierarchical Assembly
My research interests include Hierarchical Assembly, Artificial Intelligence, and Open-Endedness.